

S1-S5 breast cancer contour application#
This advanced pyXenium.contour tutorial uses the Atera Xenium WTA FFPE breast cancer export at
Y:\long\10X_datasets\Xenium\Atera\WTA_Preview_FFPE_Breast_Cancer_outs to build five HistoSeg-backed contour annotations and then run contour-native biology and topology analyses.
The five annotation classes are intentionally broader than the basic contour tutorial:
S class |
Biological compartment |
Aggregated Atera cell groups |
|---|---|---|
S1 |
Invasive tumor plus invasion-associated CAFs |
CAFs Invasive Associated, 11q13 invasive tumor, G1/S, mitotic |
S2 |
Basal-like structured DCIS plus lymphoid/DC |
Basal DCIS, dendritic cells, B cells, T lymphocytes |
S3 |
Myeloid-vascular-stromal niche |
Mast/myeloid/macrophage, CXCL14+ fibroblast, endothelial, pericyte |
S4 |
Myoepithelial/DCIS CAF/plasma compartment |
Myoepithelial, DCIS-associated CAFs, plasma cells |
S5 |
Apocrine/luminal amorphous DCIS axis |
Apocrine and luminal-like amorphous DCIS |
The committed RTD snapshot is generated from a whole-transcriptome transcript scan. The lightweight tables below are top slices from the all-gene results; no curated DEG panel is used for gene selection.
from __future__ import annotations
from pathlib import Path
import json
import shutil
import sys
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import seaborn as sns
from IPython.display import Markdown, display
def _find_repo_root() -> Path:
cwd = Path.cwd().resolve()
for candidate in (cwd, *cwd.parents):
if (candidate / 'src' / 'pyXenium').exists():
return candidate
return cwd
PROJECT_ROOT = _find_repo_root()
SRC_ROOT = PROJECT_ROOT / 'src'
if str(SRC_ROOT) not in sys.path:
sys.path.insert(0, str(SRC_ROOT))
from pyXenium.contour import (
add_contours_from_geojson,
compare_contour_cell_composition,
compare_contour_transcript_de,
generate_barrier_contour_shells,
generate_xenium_explorer_annotations,
)
from pyXenium.io import XeniumFrameChunkSource, read_xenium
from pyXenium.io.api import TRANSCRIPT_POINT_COLUMNS, TRANSCRIPT_POINT_COLUMN_TYPES
from pyXenium.io.xenium_artifacts import iter_transcript_chunks
sns.set_theme(context='notebook', style='whitegrid')
DATASET_ROOT = Path(r'Y:\long\10X_datasets\Xenium\Atera\WTA_Preview_FFPE_Breast_Cancer_outs')
HISTOSEG_ROOT = Path(r'D:\GitHub\HistoSeg')
OUTPUT_ROOT = DATASET_ROOT / 'pyxenium_tutorial_outputs' / 'atera_s1_s5_contour_application'
FIGURE_DIR = OUTPUT_ROOT / 'figures'
STATIC_SNAPSHOT_DIR = PROJECT_ROOT / 'docs' / '_static' / 'tutorials' / 'contour_s1_s5'
GRAPHCLUST_RELPATH = Path('analysis/analysis/clustering/gene_expression_graphclust/clusters.csv')
CELL_GROUPS_PATH = DATASET_ROOT / 'WTA_Preview_FFPE_Breast_Cancer_cell_groups.csv'
CELLS_PARQUET_RELPATH = Path('cells.parquet')
TRANSCRIPTS_PATH = DATASET_ROOT / 'transcripts.parquet'
S1_S5_GEOJSON = DATASET_ROOT / 'xenium_explorer_annotations.s1_s5.generated.geojson'
PIXEL_SIZE_UM = 0.2125
QUALITY_QUERY = 'quality_score >= 20 and valid == True'
RUN_HISTOSEG_EXPORT = False
RUN_TRANSCRIPT_SNAPSHOT = False
OUTPUT_ROOT.mkdir(parents=True, exist_ok=True)
FIGURE_DIR.mkdir(parents=True, exist_ok=True)
STATIC_SNAPSHOT_DIR.mkdir(parents=True, exist_ok=True)
def top_ok(frame: pd.DataFrame, n: int = 200) -> pd.DataFrame:
local = frame.loc[frame.get('status', 'ok').eq('ok')].copy()
if local.empty:
local = frame.copy()
return local.head(n).reset_index(drop=True)
def top_one_vs_rest_snapshot(frame: pd.DataFrame, per_case: int = 30) -> pd.DataFrame:
local = frame.loc[frame.get('status', 'ok').eq('ok')].copy()
if local.empty:
local = frame.copy()
return (
local.sort_values(['case', 'fdr', 'p_value', 'gene'], na_position='last', kind='stable')
.groupby('case', group_keys=False)
.head(per_case)
.reset_index(drop=True)
)
1. Build the S1-S5 structure definitions#
The official GraphClust table stores numeric cluster IDs, while the Atera cell-group CSV stores readable labels and colors. We bridge those tables and aggregate cluster IDs into the five S classes. Two user-facing names are aliased to the exact dataset labels: CAFs Invasive Associated -> CAFs, Invasive Associated and CAFs DCIS Associated -> CAFs, DCIS Associated.
S1_S5_PANEL = {
'S1': [
'CAFs Invasive Associated',
'11q13 Invasive Tumor Cells',
'11q13 Invasive Tumor Cells (G1/S)',
'11q13 Invasive Tumor Cells (Mitotic)',
],
'S2': [
'Basal-like Structured DCIS Cells',
'Dendritic Cells',
'B Cells',
'T Lymphocytes',
],
'S3': [
'Mast Cells',
'Myeloid Cells',
'Macrophages',
'CXCL14+ Fibroblasts',
'Endothelial Cells',
'Pericytes',
],
'S4': [
'Myoepithelial Cells',
'CAFs DCIS Associated',
'Plasma Cells',
],
'S5': [
'Apocrine Cells',
'Luminal-like Amorphous DCIS Cells',
],
}
LABEL_ALIASES = {
'CAFs Invasive Associated': 'CAFs, Invasive Associated',
'CAFs DCIS Associated': 'CAFs, DCIS Associated',
}
S_COLORS = {'S1': '#C43C39', 'S2': '#3F7CAC', 'S3': '#4A9D67', 'S4': '#B9802D', 'S5': '#9A5BC7'}
S_DESCRIPTIONS = {
'S1': 'Invasive tumor + CAFs',
'S2': 'Basal DCIS + lymphoid',
'S3': 'Myeloid-vascular-stromal',
'S4': 'Myoepithelial/DCIS CAF/plasma',
'S5': 'Apocrine/luminal DCIS',
}
cluster_df = pd.read_csv(DATASET_ROOT / GRAPHCLUST_RELPATH)
cell_groups_df = pd.read_csv(CELL_GROUPS_PATH).rename(columns={'cell_id': 'Barcode', 'group': 'cell_group'})
cluster_bridge = cluster_df.merge(
cell_groups_df[['Barcode', 'cell_group', 'color']],
on='Barcode',
how='inner',
)
label_to_clusters = (
cluster_bridge.groupby('cell_group')['Cluster']
.apply(lambda values: sorted(pd.Series(values).dropna().astype(int).unique().tolist()))
.to_dict()
)
label_counts = cluster_bridge.groupby('cell_group').size().to_dict()
structures = []
structure_rows = []
for idx, (structure_name, requested_labels) in enumerate(S1_S5_PANEL.items(), start=1):
cluster_ids = []
resolved_labels = []
input_cell_count = 0
for requested_label in requested_labels:
resolved_label = LABEL_ALIASES.get(requested_label, requested_label)
if resolved_label not in label_to_clusters:
raise KeyError(f'Missing Atera cell group: {requested_label!r} -> {resolved_label!r}')
cluster_ids.extend(label_to_clusters[resolved_label])
resolved_labels.append(resolved_label)
input_cell_count += int(label_counts[resolved_label])
structures.append(
{
'structure_name': structure_name,
'cluster_ids': sorted(set(cluster_ids)),
'structure_color': S_COLORS[structure_name],
'structure_id': idx,
'member_labels': tuple(resolved_labels),
}
)
structure_rows.append(
{
'structure_name': structure_name,
'description': S_DESCRIPTIONS[structure_name],
'cluster_ids': sorted(set(cluster_ids)),
'member_labels': ', '.join(resolved_labels),
'input_cell_count': input_cell_count,
}
)
structure_definition_table = pd.DataFrame(structure_rows)
display(structure_definition_table)
2. Generate HistoSeg-backed Xenium Explorer annotations#
generate_xenium_explorer_annotations(...) calls the HistoSeg multi-structure contour API and writes a Xenium Explorer-compatible GeoJSON. The legacy xenium_explorer_annotations.geojson is left untouched; this tutorial writes xenium_explorer_annotations.s1_s5.generated.geojson.
if RUN_HISTOSEG_EXPORT or not S1_S5_GEOJSON.exists():
artifacts = generate_xenium_explorer_annotations(
DATASET_ROOT,
structures=structures,
output_relpath=OUTPUT_ROOT.relative_to(DATASET_ROOT),
clusters_relpath=GRAPHCLUST_RELPATH,
cells_parquet_relpath=CELLS_PARQUET_RELPATH,
histoseg_root=HISTOSEG_ROOT if HISTOSEG_ROOT.exists() else None,
min_cells=500,
min_component_pixels=180,
xenium_pixel_size_um=PIXEL_SIZE_UM,
)
shutil.copy2(artifacts['geojson'], S1_S5_GEOJSON)
else:
artifacts = {
'geojson': str(S1_S5_GEOJSON),
'out_dir': str(OUTPUT_ROOT),
'preview_png': str(OUTPUT_ROOT / 'multi_structure_contour_preview.png'),
}
contour_counts = pd.read_csv(OUTPUT_ROOT / 's1_s5_contour_counts.csv') if (OUTPUT_ROOT / 's1_s5_contour_counts.csv').exists() else pd.read_csv(OUTPUT_ROOT / 'structure_contour_cell_counts.csv')
display(contour_counts)
artifacts
Snapshot contour counts#
The committed RTD snapshot includes this small result table, so the page remains informative even with notebook execution disabled.
structure_name |
description |
input_cell_count |
assigned_cell_count |
contour_count |
|---|---|---|---|---|
S1 |
Invasive tumor + CAFs |
72873 |
71725 |
106 |
S2 |
Basal DCIS + lymphoid |
23298 |
15935 |
358 |
S3 |
Myeloid-vascular-stromal |
26690 |
37038 |
491 |
S4 |
Myoepithelial/DCIS CAF/plasma |
33753 |
36486 |
313 |
S5 |
Apocrine/luminal DCIS |
13431 |
8861 |
28 |
Annotation preview#
The generated S1-S5 export contains multiple contours per class. In the snapshot run, the HistoSeg count table reported 106 S1, 358 S2, 491 S3, 313 S4, and 28 S5 contour components before GeoJSON polygon serialization.
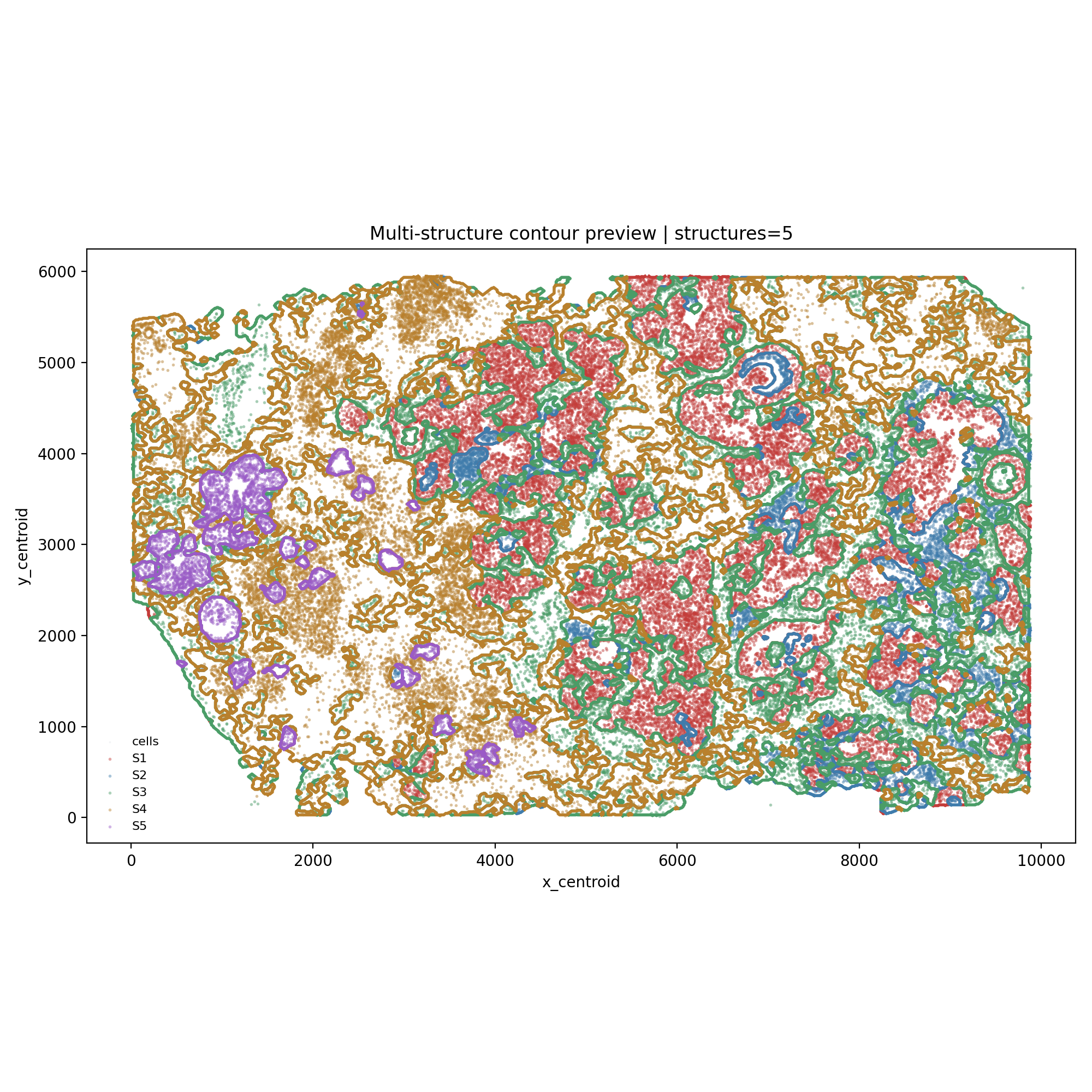
3. Load Xenium, import contours, and attach cell-group labels#
We load the cell table without materializing the full transcript parquet. When RUN_TRANSCRIPT_SNAPSHOT=True, the notebook attaches the unfiltered transcripts.parquet stream and lets compare_contour_transcript_de(..., genes=None) discover all genes from the whole transcriptome. The RTD page keeps execution off and displays the committed whole-transcriptome snapshot tables and figures.
slide = read_xenium(
str(DATASET_ROOT),
as_='slide',
include_transcripts=False,
include_boundaries=False,
include_images=False,
clusters_relpath=str(GRAPHCLUST_RELPATH),
)
cell_group_map = cell_groups_df.set_index('Barcode')['cell_group']
slide.table.obs['cell_group'] = slide.table.obs.index.map(cell_group_map).fillna('Unassigned')
add_contours_from_geojson(
slide,
S1_S5_GEOJSON,
key='s1_s5_contours',
id_key='name',
pixel_size_um=PIXEL_SIZE_UM,
)
print(slide.component_summary())
print(sorted(slide.shapes['s1_s5_contours']['assigned_structure'].dropna().unique()))
def attach_whole_transcript_source(slide, genes=None) -> None:
gene_filter = None if genes is None else set(str(gene) for gene in genes)
slide.points.pop('transcripts', None)
slide.point_sources['transcripts'] = XeniumFrameChunkSource(
columns=TRANSCRIPT_POINT_COLUMNS,
column_types=TRANSCRIPT_POINT_COLUMN_TYPES,
chunk_iter_factory=lambda: iter_transcript_chunks(str(TRANSCRIPTS_PATH), genes=gene_filter),
attrs={
'source_path': str(TRANSCRIPTS_PATH),
'gene_filter': None if gene_filter is None else tuple(sorted(gene_filter)),
'analysis_mode': 'whole_transcriptome',
},
preserve_extra_columns=True,
)
if RUN_TRANSCRIPT_SNAPSHOT:
attach_whole_transcript_source(slide)
4. Transcript-level contour DEG#
compare_contour_transcript_de(...) aggregates raw transcript counts into contour-level pseudobulks, computes contour-level CPM/log1p CPM, and treats each contour as a replicate. Here genes=None means the DEG scan is whole-transcriptome: every gene observed in the unfiltered transcript stream is tested before the top snapshot rows are selected for RTD. For this whole-transcriptome screening step, assignment='cell_id' uses the cell_id field in transcripts.parquet and assigns cell-associated transcripts through the contour membership of each cell centroid, which is much faster than point-in-polygon testing 740M transcript coordinates.
if RUN_TRANSCRIPT_SNAPSHOT:
deg_result_full = compare_contour_transcript_de(
slide,
contour_key='s1_s5_contours',
groupby='assigned_structure',
genes=None,
assignment='cell_id',
comparisons=('global', 'one_vs_rest'),
transcript_query=QUALITY_QUERY,
min_total_transcripts_per_contour=1,
min_contours_per_group=2,
include_zero_counts=False,
return_contour_gene=False,
)
deg_result_full['global_de'].to_csv(
OUTPUT_ROOT / 's1_s5_transcript_de_global.whole_transcriptome.full.csv', index=False
)
deg_result_full['one_vs_rest_de'].to_csv(
OUTPUT_ROOT / 's1_s5_transcript_de_one_vs_rest.whole_transcriptome.full.csv', index=False
)
deg_result = {
'global_de': top_ok(deg_result_full['global_de'], 200),
'one_vs_rest_de': top_one_vs_rest_snapshot(deg_result_full['one_vs_rest_de'], per_case=30),
}
deg_result['global_de'].to_csv(OUTPUT_ROOT / 's1_s5_transcript_de_global.csv', index=False)
deg_result['one_vs_rest_de'].to_csv(OUTPUT_ROOT / 's1_s5_transcript_de_one_vs_rest.csv', index=False)
else:
deg_result = {
'global_de': pd.read_csv(STATIC_SNAPSHOT_DIR / 's1_s5_transcript_de_global.csv'),
'one_vs_rest_de': pd.read_csv(STATIC_SNAPSHOT_DIR / 's1_s5_transcript_de_one_vs_rest.csv'),
}
display(deg_result['global_de'].head(12))
display(deg_result['one_vs_rest_de'].head(12))
Snapshot DEG tables#
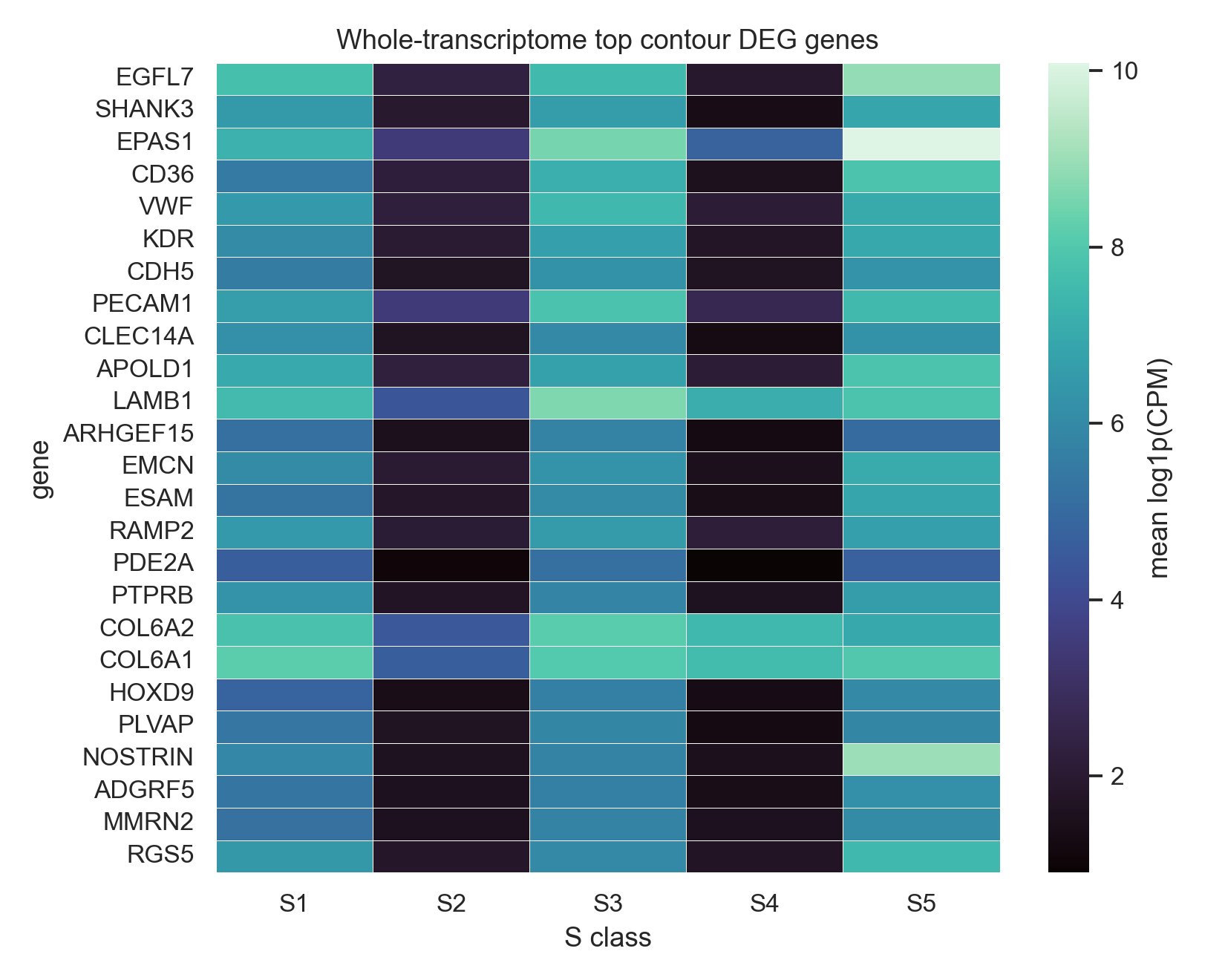
Top global genes from the whole-transcriptome contour-level Kruskal-Wallis test. These rows are a committed top slice from 18,028 tested genes, not a curated panel.
gene |
top_group |
bottom_group |
delta_log1p_cpm |
delta_mean_density_per_um2 |
fdr |
|---|---|---|---|---|---|
EGFL7 |
S3 |
S4 |
3.47 |
0.00167 |
6.01e-88 |
SHANK3 |
S3 |
S4 |
3.17 |
0.00148 |
2.48e-73 |
EPAS1 |
S3 |
S2 |
3.08 |
0.004026 |
2.48e-73 |
CD36 |
S3 |
S4 |
3.35 |
0.00161 |
3.09e-73 |
VWF |
S3 |
S4 |
3.48 |
0.003595 |
2.02e-71 |
KDR |
S3 |
S4 |
3.01 |
0.001556 |
9.05e-67 |
CDH5 |
S3 |
S2 |
2.77 |
0.0009097 |
1.02e-64 |
PECAM1 |
S3 |
S4 |
3.16 |
0.002475 |
8.99e-64 |
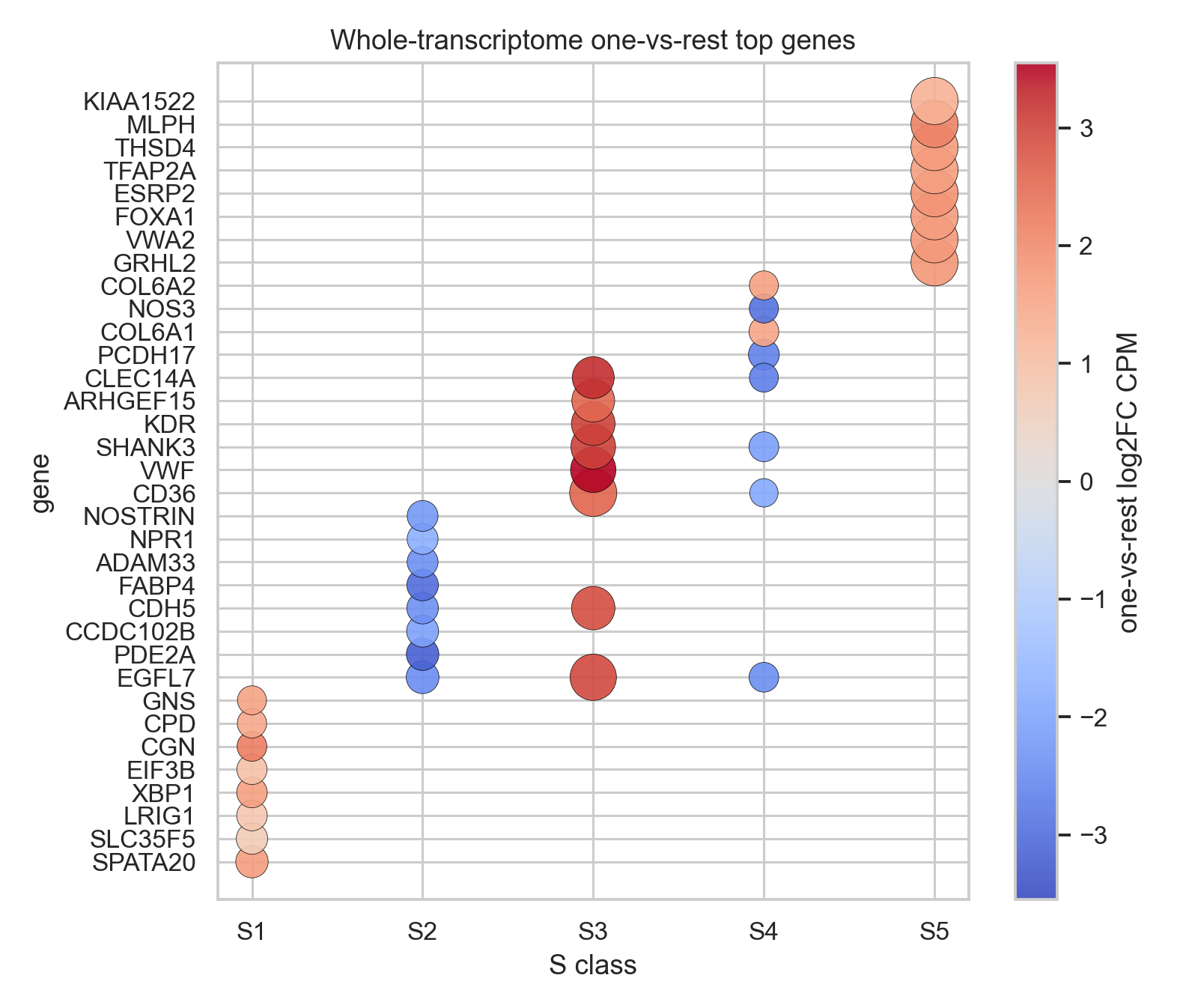
Top two one-vs-rest whole-transcriptome genes per S class:
case |
gene |
log2fc_cpm |
fdr |
|---|---|---|---|
S1 |
SPATA20 |
1.71 |
5.98e-36 |
S1 |
SLC35F5 |
0.68 |
2.03e-33 |
S2 |
EGFL7 |
-2.44 |
9.62e-37 |
S2 |
PDE2A |
-3.19 |
4.2e-36 |
S3 |
CD36 |
2.6 |
3.62e-82 |
S3 |
EGFL7 |
2.97 |
4.46e-78 |
S4 |
PCDH17 |
-2.64 |
4.86e-32 |
S4 |
SHANK3 |
-2.11 |
7.78e-30 |
S5 |
GRHL2 |
1.83 |
1.73e-175 |
S5 |
VWA2 |
1.77 |
2.61e-156 |
DEG visualization#
The heatmap shows mean contour-level log1p(CPM) for top genes selected from the whole-transcriptome global test. The dotplot summarizes the strongest whole-transcriptome one-vs-rest signals per S class.
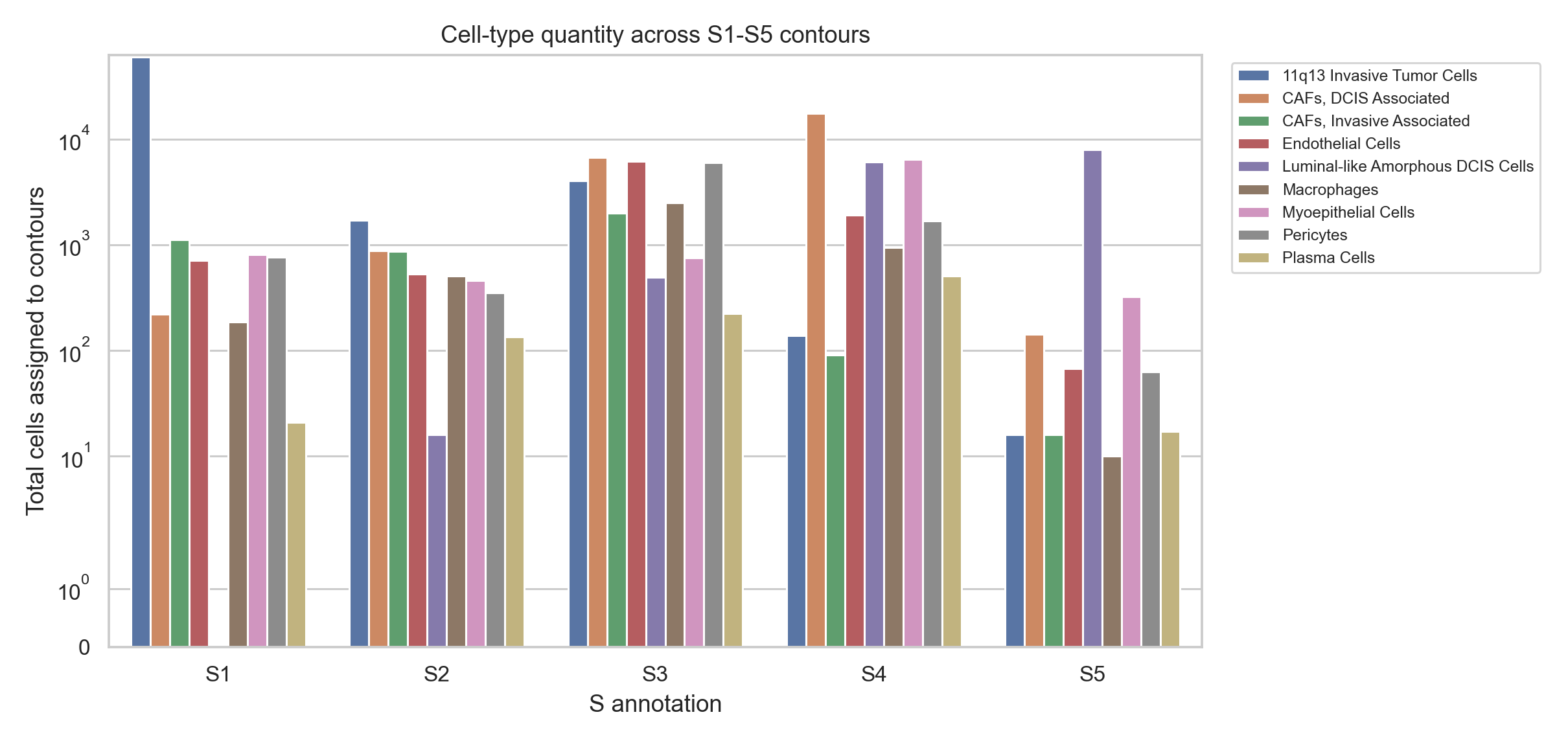
5. Cell-type composition across contour classes#
compare_contour_cell_composition(...) assigns cell centroids to every contour independently. It reports both proportions and quantities, then tests S-class differences with Kruskal-Wallis globally and Mann-Whitney tests pairwise.
if RUN_TRANSCRIPT_SNAPSHOT:
cell_result = compare_contour_cell_composition(
slide,
contour_key='s1_s5_contours',
groupby='assigned_structure',
cell_type_key='cell_group',
)
cell_result['group_summary'].to_csv(OUTPUT_ROOT / 's1_s5_cell_composition_group_summary.csv', index=False)
pd.concat(
[
cell_result['global_stats'].assign(test_scope='global'),
cell_result['pairwise_stats'].assign(test_scope='pairwise'),
],
ignore_index=True,
sort=False,
).to_csv(OUTPUT_ROOT / 's1_s5_cell_composition_stats.csv', index=False)
else:
cell_result = {
'group_summary': pd.read_csv(STATIC_SNAPSHOT_DIR / 's1_s5_cell_composition_group_summary.csv'),
'stats': pd.read_csv(STATIC_SNAPSHOT_DIR / 's1_s5_cell_composition_stats.csv'),
}
display(cell_result['group_summary'].head(12))
if 'stats' in cell_result:
display(cell_result['stats'].head(12))
Snapshot cell-composition statistics#
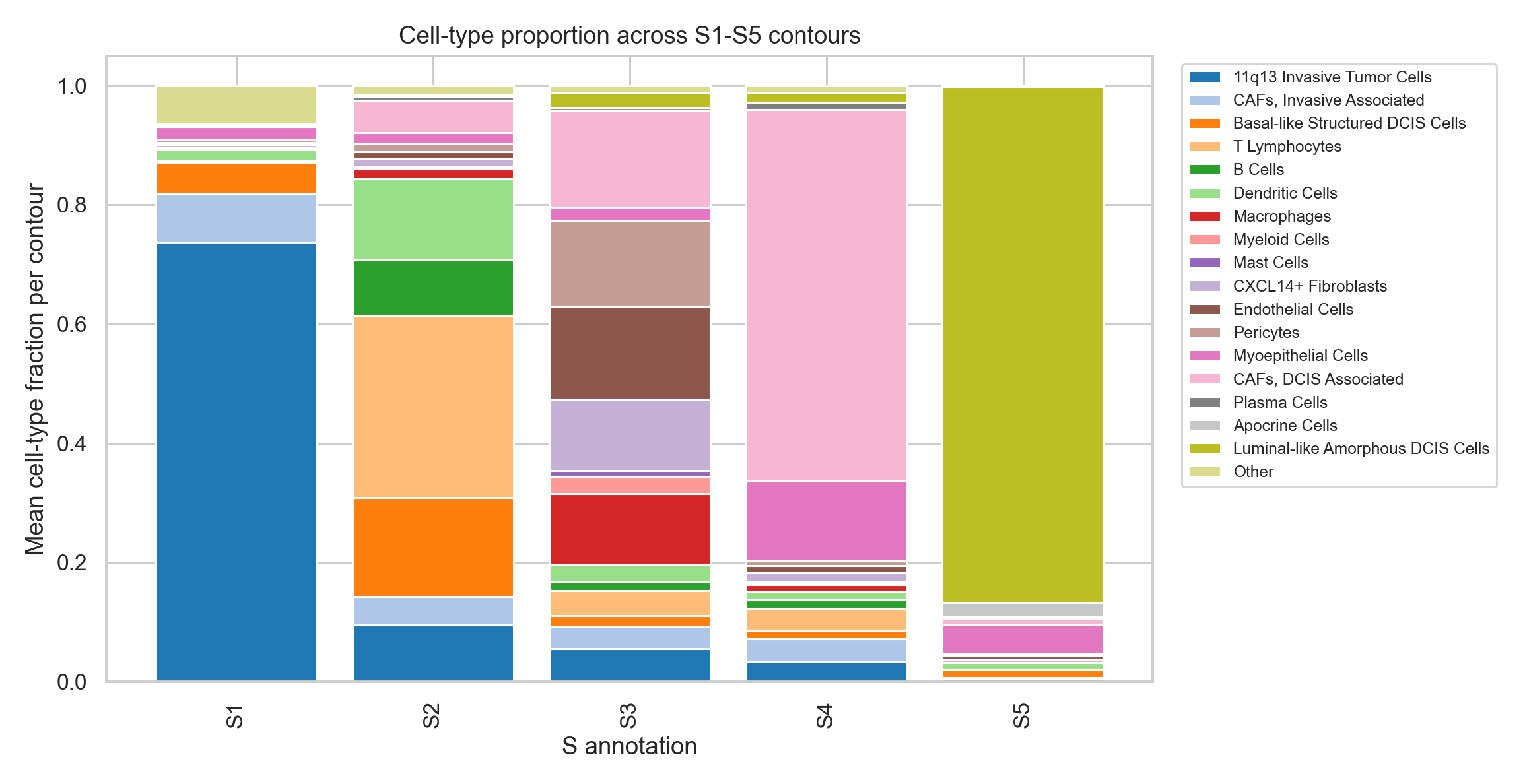
Top global cell-type composition differences across S1-S5 contours:
cell_type |
metric |
top_group |
bottom_group |
fdr |
|---|---|---|---|---|
Pericytes |
n_cells |
S3 |
S2 |
6.01e-98 |
Endothelial Cells |
n_cells |
S3 |
S2 |
1.40e-94 |
Pericytes |
fraction |
S3 |
S5 |
3.49e-90 |
Endothelial Cells |
fraction |
S3 |
S1 |
1.06e-89 |
Luminal-like Amorphous DCIS Cells |
n_cells |
S5 |
S1 |
5.84e-77 |
CXCL14+ Fibroblasts |
n_cells |
S1 |
S2 |
5.35e-76 |
CAFs, DCIS Associated |
fraction |
S4 |
S1 |
7.76e-70 |
Luminal-like Amorphous DCIS Cells |
fraction |
S5 |
S1 |
2.62e-67 |
Composition visualization#
6. Barrier-aware S1/S4/S5 boundary shells#
The boundary question asks how S1, S4, and S5 contours behave when expanded inward and outward in 20 um steps. We generate 10 signed rings: [-100,-80] ... [-20,0] and [0,20] ... [80,100]. Outward rings are clipped by all other S1-S5 contour interiors, so a shell stops contributing when it reaches another annotated contour.
selected_frame = slide.shapes['s1_s5_contours'].loc[
slide.shapes['s1_s5_contours']['assigned_structure'].isin(['S1', 'S4', 'S5'])
].copy()
slide.shapes['s1_s4_s5_contours'] = selected_frame
slide.metadata.setdefault('contours', {})['s1_s4_s5_contours'] = {
'units': 'micron',
'derived_from_key': 's1_s5_contours',
}
if 's1_s4_s5_barrier_shells' not in slide.shapes:
generate_barrier_contour_shells(
slide,
contour_key='s1_s4_s5_contours',
barrier_contour_key='s1_s5_contours',
inward=100.0,
outward=100.0,
step_size=20.0,
output_key='s1_s4_s5_barrier_shells',
)
shell_frame = slide.shapes['s1_s4_s5_barrier_shells']
slide.shapes['s1_s4_s5_boundary_outward_0_60'] = shell_frame.loc[
shell_frame['shell_direction'].eq('outward')
& (shell_frame['shell_start'] >= 0)
& (shell_frame['shell_end'] <= 60)
].copy()
slide.metadata.setdefault('contours', {})['s1_s4_s5_boundary_outward_0_60'] = {
'units': 'micron',
'derived_from_key': 's1_s4_s5_barrier_shells',
}
display(shell_frame[['contour_id', 'source_contour_id', 'assigned_structure', 'shell_start', 'shell_end', 'shell_direction']].head())
print('shell contours:', shell_frame['contour_id'].nunique())
print('outward 0-60 um shells:', slide.shapes['s1_s4_s5_boundary_outward_0_60']['contour_id'].nunique())
if RUN_TRANSCRIPT_SNAPSHOT:
boundary_result_full = compare_contour_transcript_de(
slide,
contour_key='s1_s4_s5_boundary_outward_0_60',
groupby='assigned_structure',
genes=None,
assignment='cell_id',
comparisons=('global',),
transcript_query=QUALITY_QUERY,
min_total_transcripts_per_contour=0,
min_contours_per_group=2,
include_zero_counts=False,
return_contour_gene=False,
)
boundary_stats = top_ok(boundary_result_full['global_de'], 200)
boundary_result_full['global_de'].to_csv(
OUTPUT_ROOT / 's1_s4_s5_boundary_de_global_outward_0_60.whole_transcriptome.full.csv', index=False
)
boundary_stats.to_csv(OUTPUT_ROOT / 's1_s4_s5_boundary_de_global_outward_0_60.csv', index=False)
top_boundary_genes = boundary_stats['gene'].head(10).tolist()
attach_whole_transcript_source(slide, genes=top_boundary_genes)
shell_result = compare_contour_transcript_de(
slide,
contour_key='s1_s4_s5_barrier_shells',
groupby='assigned_structure',
genes=top_boundary_genes,
comparisons=('global',),
transcript_query=QUALITY_QUERY,
min_total_transcripts_per_contour=0,
min_contours_per_group=2,
include_zero_counts=True,
return_contour_gene=True,
)
shell_gene = shell_result['contour_gene'].copy()
boundary_curves = (
shell_gene.loc[shell_gene['gene'].isin(top_boundary_genes)]
.groupby(['assigned_structure', 'gene', 'shell_start', 'shell_end', 'shell_mid'], as_index=False)
.agg(
mean_density_per_um2=('density_per_um2', 'mean'),
sem_density_per_um2=('density_per_um2', lambda values: float(pd.Series(values).sem())),
n_shells=('contour_id', 'nunique'),
total_count=('count', 'sum'),
)
.sort_values(['gene', 'assigned_structure', 'shell_mid'], kind='stable')
)
boundary_curves.to_csv(OUTPUT_ROOT / 's1_s4_s5_barrier_ring_density_top10.csv', index=False)
else:
boundary_curves = pd.read_csv(STATIC_SNAPSHOT_DIR / 's1_s4_s5_barrier_ring_density_top10.csv')
boundary_stats = pd.read_csv(STATIC_SNAPSHOT_DIR / 's1_s4_s5_boundary_de_global_outward_0_60.csv')
display(boundary_stats.head(10))
display(boundary_curves.head(10))
Snapshot boundary DE table#
Top boundary genes were selected from a whole-transcriptome DEG scan on outward 0-60 um barrier-clipped shells. The screening step uses cell-associated transcripts assigned by cell centroid for scalability; the density curves below re-count these top genes from transcript coordinates across all 10 signed-distance steps.
gene |
top_group |
bottom_group |
delta_log1p_cpm |
delta_mean_density_per_um2 |
fdr |
|---|---|---|---|---|---|
SLC39A6 |
S1 |
S4 |
1.48 |
0.00204 |
2.81e-21 |
AGR3 |
S1 |
S4 |
1.56 |
0.003781 |
2.81e-21 |
CERS2 |
S1 |
S4 |
1.57 |
0.001149 |
2.81e-21 |
TPBG |
S1 |
S5 |
1.07 |
0.0009283 |
2.81e-21 |
TJP1 |
S1 |
S4 |
1.16 |
0.0006458 |
2.91e-21 |
ELAPOR1 |
S1 |
S4 |
1.65 |
0.002875 |
2.98e-21 |
DDB1 |
S1 |
S4 |
1.53 |
0.001435 |
4.46e-21 |
GNS |
S5 |
S4 |
2.03 |
0.01711 |
4.49e-21 |
TNFSF10 |
S1 |
S4 |
1.68 |
0.004175 |
4.49e-21 |
SPATA20 |
S1 |
S4 |
1.52 |
0.002 |
5.14e-21 |
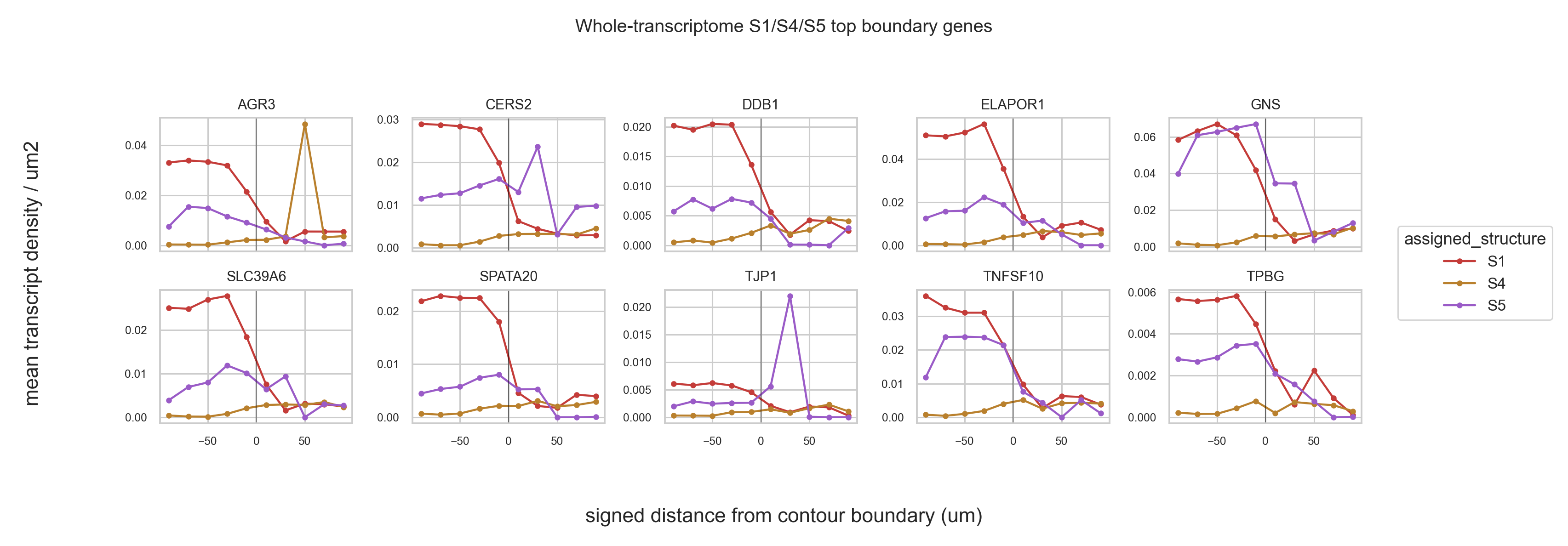
Boundary density curves#
Top boundary genes were selected from a whole-transcriptome DEG scan on cell-associated transcripts whose cell centroids fall in the outward 0-60 um barrier-clipped shells. After those top genes are selected, their density curves are re-counted from transcript coordinates across the full 10 signed-distance steps. Negative distances are inside the source contour; positive distances are outside but barrier-clipped so they do not enter another contour.
7. Biological interpretation#
The S1-S5 contours recover expected compartment-level organization, but the whole-transcriptome scan also makes the dominant axes more explicit than the earlier marker-panel snapshot.
S1 remains an invasive tumor/CAF-rich compartment. Its one-vs-rest top genes include
SPATA20,SLC35F5,LRIG1,XBP1,CGN,CPD,GNS, andANXA2, consistent with an epithelial tumor program mixed with stress/secretory and invasion-associated biology.S2 is a basal-like structured DCIS plus lymphoid/DC compartment. In the whole-transcriptome one-vs-rest table, its strongest contrasts are largely negative vascular/endothelial genes such as
EGFL7,PDE2A,CCDC102B,CDH5, andFABP4, supporting that S2 is less vascular than the S3/S5-rich microenvironments.S3 is the clearest vascular-stromal niche. The global top genes are dominated by endothelial/perivascular markers, including
EGFL7,EPAS1,VWF,KDR,CDH5,PECAM1,CLEC14A,EMCN,ESAM,RAMP2,PTPRB,PLVAP,MMRN2, andRGS5.S4 combines myoepithelial cells, DCIS-associated CAFs, and plasma cells. Its strongest one-vs-rest contrasts include matrix-associated
COL6A1andCOL6A2, while several endothelial genes are lower than the rest, separating it from the S3 vascular niche.S5 is the luminal/apocrine DCIS axis. Its one-vs-rest top genes include epithelial/luminal regulatory and differentiation genes such as
MLPH,TFAP2A,ESRP2,FOXA1,GRHL2, andVWA2.For S1/S4/S5 boundary ecology, whole-transcriptome outward 0-60 um screening selected
SLC39A6,AGR3,CERS2,TPBG,TJP1,ELAPOR1,DDB1,GNS,TNFSF10, andSPATA20. Coordinate-based density curves show many of these genes are highest inside S1 and/or S5 and decline across the boundary, which supports an epithelial/luminal boundary program rather than a purely stromal outward signal.
A practical analysis pattern emerges: use whole-transcriptome compare_contour_transcript_de to identify contour-level programs, compare_contour_cell_composition to explain which cell types make those programs plausible, and generate_barrier_contour_shells plus coordinate-counted transcript density curves to test whether a program sits inside the contour, peaks at the boundary, or extends into the surrounding microenvironment.
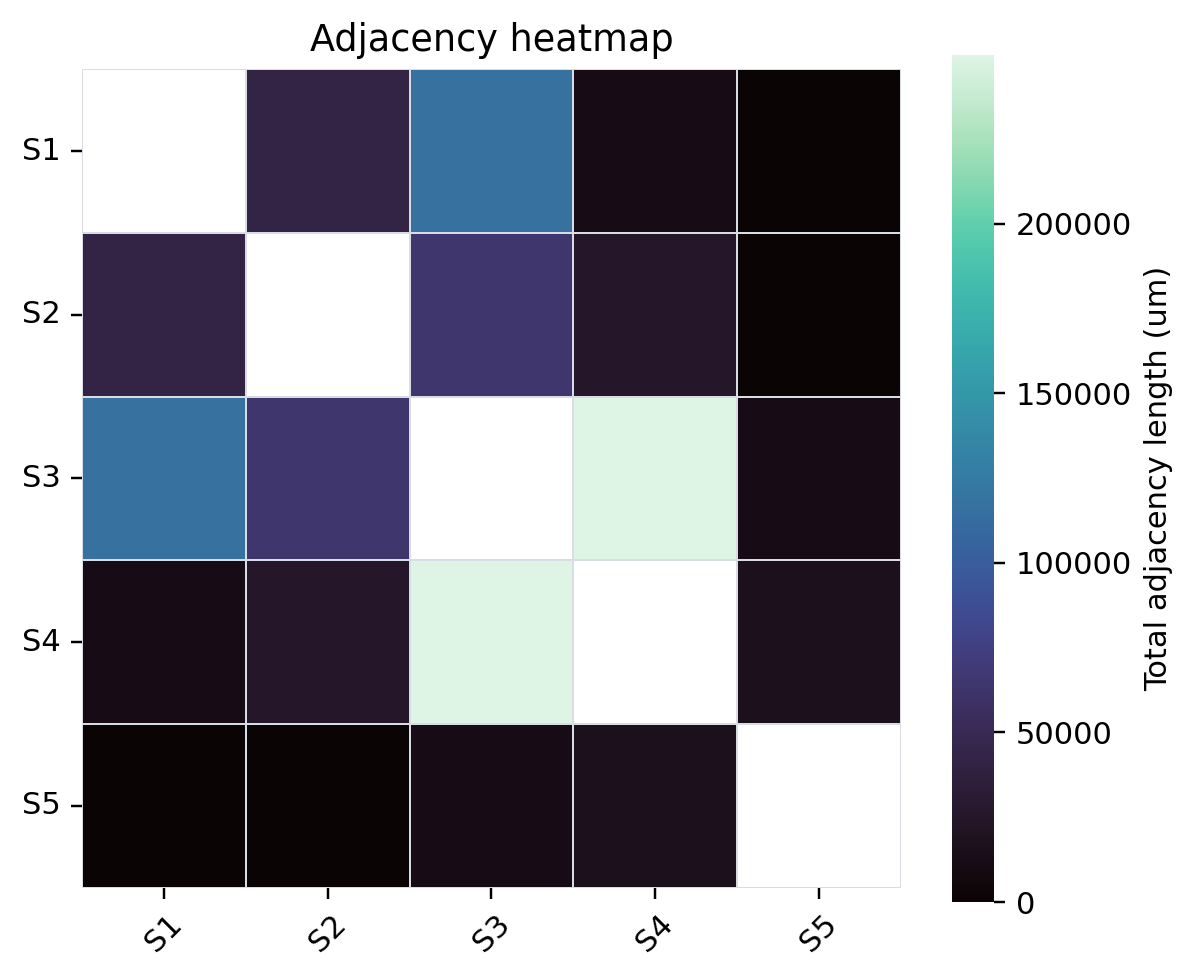
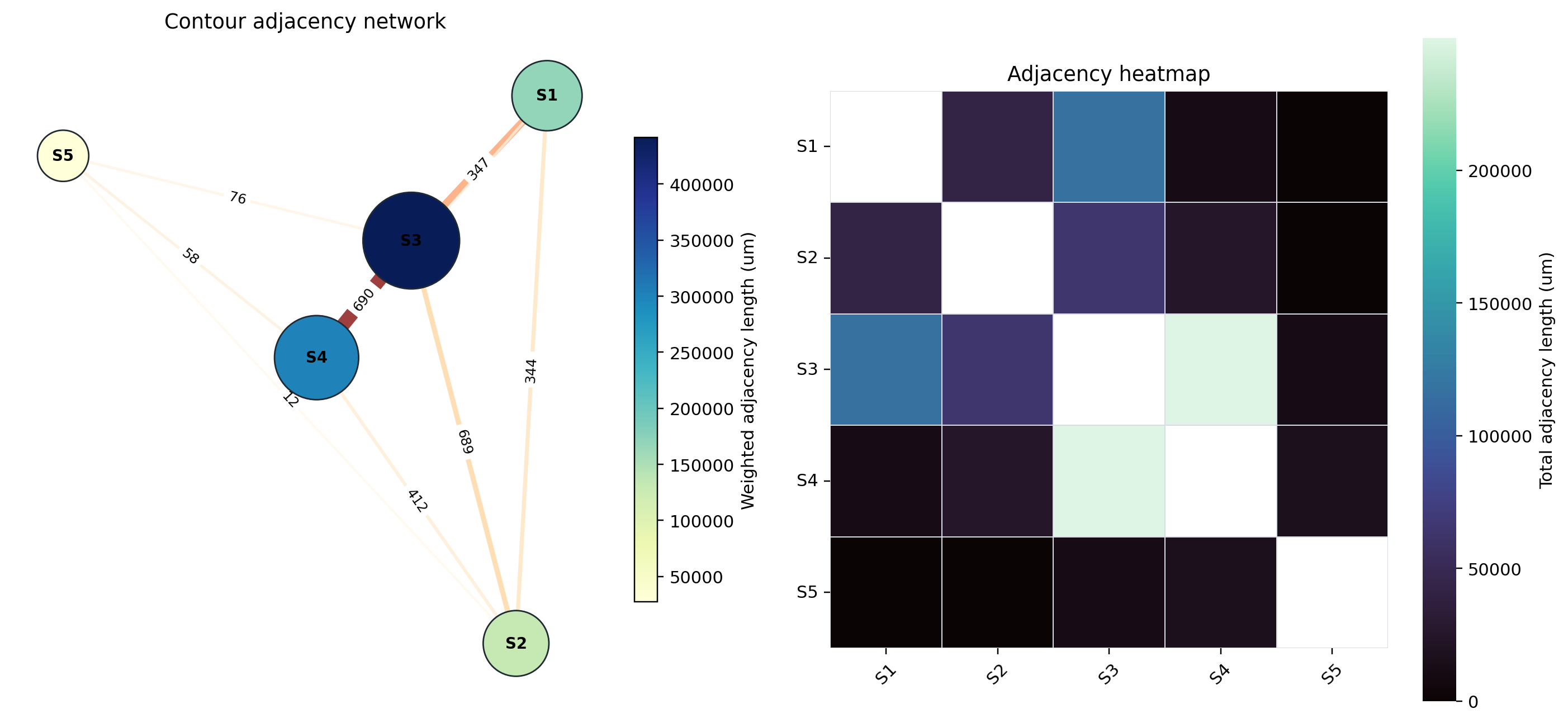
8. Contour adjacency network analysis#
HistoSeg also provides a contour adjacency network layer. Here we reuse the already imported S1-S5 contour geometries, aggregate pairwise boundary-neighbor and enclosure relationships by assigned_structure, and render a compact network/heatmap snapshot for RTD. The adjacency score combines shared border length with enclosure-derived inner contour perimeter, so it captures both geography-like border contact and one compartment wrapping around another.
histoseg_src = HISTOSEG_ROOT / 'src'
if histoseg_src.exists() and str(histoseg_src) not in sys.path:
sys.path.insert(0, str(histoseg_src))
from histoseg.contour import ContourAdjacencyConfig, run_contour_adjacency
from pyXenium.contour._geometry import contour_frame_to_geometry_table
ADJACENCY_ROOT = OUTPUT_ROOT / 's1_s5_contour_adjacency'
ADJACENCY_ROOT.mkdir(parents=True, exist_ok=True)
adjacency_contours = contour_frame_to_geometry_table(
slide.shapes['s1_s5_contours'],
contour_key='s1_s5_contours',
)[['contour_id', 'assigned_structure', 'classification_name', 'structure_id', 'geometry']].copy()
adjacency_contours = adjacency_contours.rename(columns={'assigned_structure': 'structure'})
adjacency_result = run_contour_adjacency(
ContourAdjacencyConfig(
contours=adjacency_contours,
out_dir=ADJACENCY_ROOT,
groupby='structure',
contour_id_col='contour_id',
geometry_col='geometry',
boundary_tolerance=1.0,
min_shared_boundary=0.0,
enclosure_min_fraction=0.95,
dpi=220,
save_preview_png=True,
)
)
adjacency_edges = pd.read_csv(adjacency_result.edges_csv)
adjacency_matrix = pd.read_csv(adjacency_result.matrix_csv, index_col=0)
snapshot_files = {
adjacency_result.edges_csv: STATIC_SNAPSHOT_DIR / 's1_s5_contour_adjacency_edges.csv',
adjacency_result.matrix_csv: STATIC_SNAPSHOT_DIR / 's1_s5_contour_adjacency_matrix.csv',
adjacency_result.network_png: STATIC_SNAPSHOT_DIR / 's1_s5_contour_adjacency_network.png',
adjacency_result.heatmap_png: STATIC_SNAPSHOT_DIR / 's1_s5_contour_adjacency_heatmap.png',
adjacency_result.combined_png: STATIC_SNAPSHOT_DIR / 's1_s5_contour_adjacency_overview.png',
}
for src, dst in snapshot_files.items():
if src is not None:
shutil.copy2(src, dst)
display(adjacency_edges.sort_values('total_adjacency_length_um', ascending=False).head(10))
display(adjacency_matrix)
adjacency_result
Snapshot adjacency edge table#
The committed RTD snapshot below was generated with HistoSeg cca55df using boundary_tolerance=1.0 um and enclosure_min_fraction=0.95. It summarizes 1578 S1-S5 contours into 10 cross-structure group pairs; 9 pairs have non-zero adjacency.
group_a |
group_b |
boundary_pair_count |
boundary_shared_length_um |
enclosure_pair_count |
enclosure_inner_boundary_length_um |
total_adjacency_length_um |
|---|---|---|---|---|---|---|
S3 |
S4 |
557 |
169686 |
133 |
80123 |
249809 |
S1 |
S3 |
323 |
101744 |
24 |
14576.8 |
116321 |
S2 |
S3 |
672 |
61709.2 |
17 |
2671.4 |
64380.6 |
S1 |
S2 |
319 |
33515.6 |
25 |
8347.4 |
41863 |
S2 |
S4 |
374 |
20373.1 |
38 |
4923 |
25296.1 |
S4 |
S5 |
43 |
6750.1 |
15 |
9422.4 |
16172.5 |
S1 |
S4 |
163 |
10859.2 |
1 |
61.1 |
10920.2 |
S3 |
S5 |
75 |
10650.1 |
1 |
266.9 |
10917 |
S2 |
S5 |
12 |
554.3 |
0 |
0 |
554.3 |
Adjacency network and heatmap#
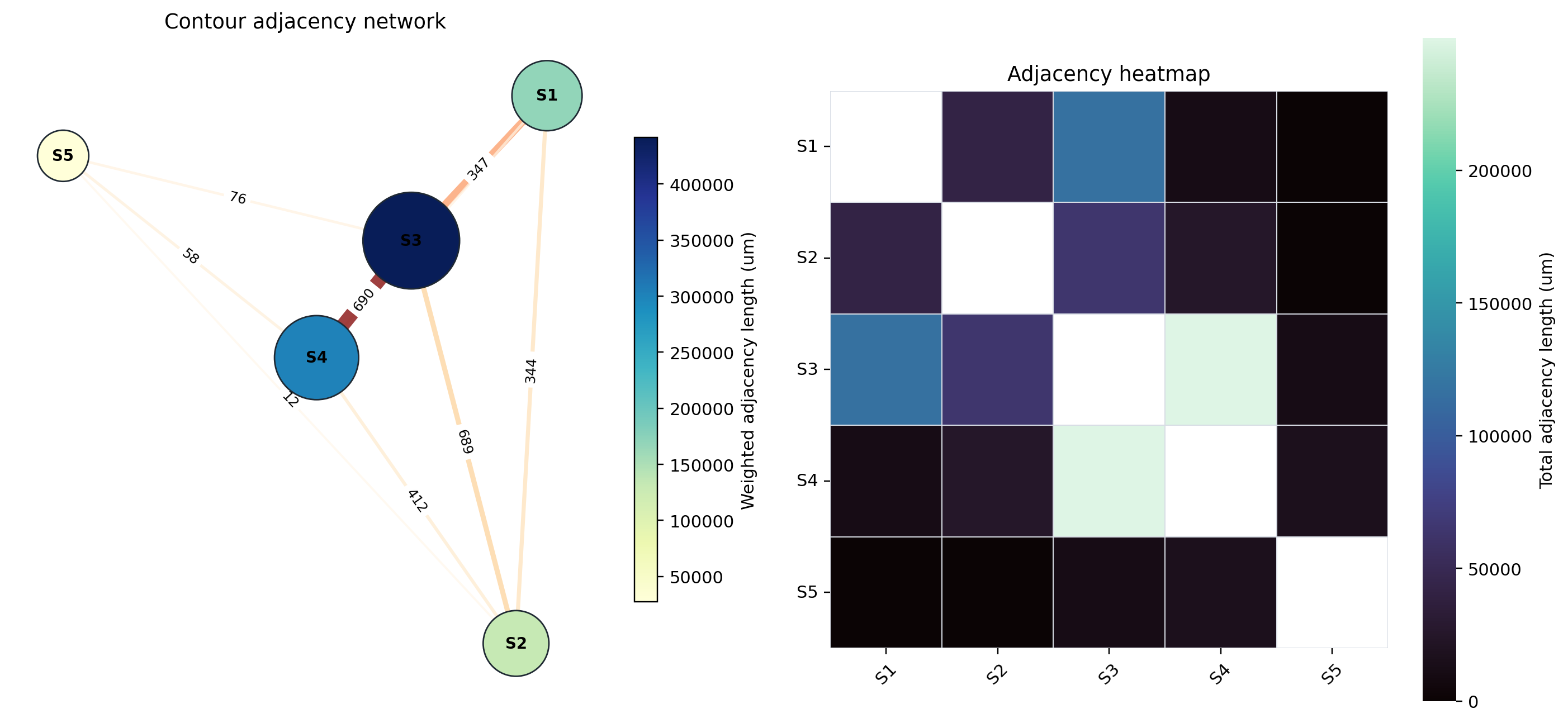
The network emphasizes the same geography seen in the table. S3 and S4 form the dominant adjacency axis (249809 um total adjacency), reflecting extensive contact between the myeloid-vascular-stromal niche and the myoepithelial/DCIS CAF/plasma compartment. S1 is most strongly connected to S3 (116321 um), supporting an invasive tumor/CAF boundary with vascular-myeloid stroma. S2 also contacts S3 (64381 um), while S5 is comparatively peripheral and has no direct positive adjacency with S1 in this S1-S5 contour export.
For reproducibility, the adjacency matrix in microns is:
S1 |
S2 |
S3 |
S4 |
S5 |
|
|---|---|---|---|---|---|
S1 |
nan |
41863 |
116321 |
10920.2 |
0 |
S2 |
41863 |
nan |
64380.6 |
25296.1 |
554.3 |
S3 |
116321 |
64380.6 |
nan |
249809 |
10917 |
S4 |
10920.2 |
25296.1 |
249809 |
nan |
16172.5 |
S5 |
0 |
554.3 |
10917 |
16172.5 |
nan |
9. Output files#
The lightweight RTD snapshot uses these committed top-slice DEG artifacts plus the compact HistoSeg adjacency outputs:
{kind=link}
{kind=link}
{kind=link}
Local reruns also write full all-gene DEG tables and the full adjacency run directory under pyxenium_tutorial_outputs/atera_s1_s5_contour_application/ beside the Xenium export. Those full tables are intentionally not committed to the docs repository.